Hybrid Molecular Nano Frameworks for Disease Modeling Using Neuro Symbolic Machine Learning
Authors :
Amanlou Ismail Salih Biswas
Address :
Research Center for Cyber Security, Faculty of Information Science and Technology, Universiti Kebangsaan Malaysia, Bangi 43600, Selangor, Malaysia
Department of Computer Engineering, Lebanese French University, Kurdistan Region, Iraq
Abstract :
Disease modelling has advanced significantly when computational methods and molecular data are used. For prediction accuracy and interpretability, this initiative proposes to create a hybrid molecular nano framework for disease modelling employing neuro-symbolic machine learning, with Graph Attention Networks (GAT) as the central method. The GAT depicts molecules as graphs, and the technique interactively assigns focus to atom-bond interactions to extract structural and relational properties. These traits' anticipated disease-relevant relationships and biological objectives are paired with symbolic reasoning, which uses molecular similarity metrics (such as Tanimoto coefficients) and 3D structure data. One of the main discoveries is the successful use of GAT to capture necessary chemical substructures and achieve better prediction performance than conventional models. The hybrid framework showed excellent interpretability, effectively connecting biological targets like CYP2D6 and HERG to molecular patterns and offering insights into disease processes. Finally, combining GAT with symbolic reasoning reveals a viable strategy for molecula
Keywords :
Disease modelling, Neuro-symbolic machine learning, Molecular similarity, interpretability, Graph Attention Networks.
1.Introduction
Integrating disease modelling, molecular data analysis, and computational approaches has converted to the biomedical research area. Over the last few decades, molecular modelling, artificial intelligence, and machine learning developments have created new opportunities to comprehend the intricate connections between disease manifestations and molecular structures [1, 2]. When incorporated into comprehensive mathematical algorithms, the extensive molecular datasets that academics presently are entitled to—such as genomic, proteomic, and structural data—can offer novel knowledge of disease leads, therapeutic targets, and prospects for solutions [3, 4]. However, despite this progress, numerous challenges exist to overcome to develop exact and comprehensible prediction models
Molecular methods must accurately represent and analyse their physical and relational features [5]. Traditional computational techniques frequently fail when overcoming the disparity between comprehension and predictive accuracy. Neural networks work based on deep learning, and other black-box algorithms have excellent accuracy in forecasting. Still, their lack of transparency prevents them from being widely used in crucial areas like personalised healthcare and drug development [6]. In contrast, readability is offered by governed-by-rules, symbolic thinking models. Still, they frequently fail to provide the capacity and versatility required to verify massive amounts of molecular information [7]. By bringing forward a hybrid molecular nano-framework that integrates Graph Attention Networks (GAT) with neurosymbolic machine learning, the present research aims to overcome these constraints.
Since complex compounds can be viewed as networks with elements as vertices and molecular connections as borders, Graph Attention Networks (GATs) have applications for simulating molecular mechanisms [8]. When paired with symbolic logic, which uses 3D structure data and similarity parameters (such as Tanimoto coefficients), GATs can more accurately and interpretably forecast correlations.
The main contributions of this study:
- The hybrid molecular nano-framework Development of a Hybrid Framework successfully balances interpretability and prediction accuracy in disease modelling by fusing GAT with symbolic reasoning.
- A better understanding of molecule-disease relationships is possible because of the enhanced molecular representation GATs model, which incorporates crucial chemical substructures and relational patterns.
- By creating significant relationships among molecular patterns and biological targets like CYP2D6 and HERG, biological insight and interpretability provide insights into the processes underlying illness.
The rest of the paper is as follows: The current state of work is given in Section 2, which includes an explanation of the investigation gap. The suggested approach is explained in Section 3, with the hybrid framework's structure and its integration of GAT and symbolic analysis. The study layout, datasets, and measurement evaluations are laid out in Section 4. The primary findings are addressed in Section 5, which compares the outcomes with the most advanced models and emphasises how comprehensible predictions are. A review of the contributions, possible uses, and future study targets can be found in Section 6, which brings the investigation to a close.
2. Literature Survey
S. Xia et al.[9]The research involves innovative algorithms for computation, including deep neural networks for predicting estimated and exploratory molecular characteristics through atomic mechanics-optimised patterns, delta neural network function scores for proteinligand connecting as well as simulated screening, and AlphaSpace for pocket-guided rational development focusing on protein-protein interactions. The information sets incorporate actual molecule property data, large drug libraries, and protein-protein interaction architectures. The FDA-approved kinase inhibitor Erlotinib was used as a successful validation case. The findings showed increased efficiency in virtual screening, greater accuracy in docking scores, and trustworthy predictions of molecular features. Limitations still exist, though, such as difficulties with model generalizability across many molecular systems, precision in predicting new interactions, and high computing resource requirements for large-scale molecular simulations.
J. Pinto et al.[10] Employing the ADAM optimal method, probabilistic normalisation, and semidirect sensibility formulas for training, the provided analytical architecture integrates molecular models with deep neural networks in compliance with the SBML standard. Specifically, the P58IPK signal transduction model, the yeast glycolytic oscillations approach, and the bacteria Escherichia coli alanine synthesising model are among the publicly accessible SBML models in the collection. The findings demonstrate better efficiency and versatility in the combination of model simulations, allowing more thorough analysis and interaction with current SBML databases. However, drawbacks include difficulties scaling hybrid models for highly complex biological systems and possible computing costs.
N. A. Aljarallah et al.[11] methods for searching through online databases and rigorous selection standards, this research thoroughly investigates machine learning (ML) algorithms for neurological medical diagnosis that depend on genes and chemical processes. Data collection encompasses 24 meticulously selected research that evaluates techniques and outcomes while concentrating on different neurological illnesses. The research results demonstrate that efficient predictive models may implement customised therapies, employing chromosomal and molecular information to enhance treatment plans and diagnostic precision. Nevertheless, there are still issues with model scalability, generalisation, and integration into healthcare environments.
C. Stavrogiannis et al. [12] The present research predicts the density and the thermal conductivity of Ar, Kr, Xe, O, and N in a range of liquid states, encompassing gas, liquid, and supersonic circumstances, using molecular dynamics (MD) models and nine machine learning (ML) techniques. The dataset is expanded using MD simulations to encompass a wider pressure-temperature (P-T) range after being generated through experimental research sources. Results show excellent accuracy in predicting, and tree-based machine learning architectures outperform conventional trials and computationally demanding simulations. Fortunately, drawbacks include the need for superior input information to provide accurate forecasts and the possibility of performance declines under challenging conditions.
H. Narayanan et al. [13] To enhance chromatography capturing procedures, this paper provides several hybrid designs that align across a "degree of hybridisation" axis and combine data-driven and mechanistic techniques. The collection comprises chromatography procedure data from experiments evaluated at various inversion degrees. The findings demonstrate that mixed approaches perform better in terms of accuracy of predictions, extension skills, process comprehension, and practical application than strictly physiological or data-driven approaches.
Zhao et al.[14] Employing Hybrid Units (HUs) as gateways for adaptable and effective transfer of data, this research suggests a Hybrid Neural Network (HNN) structure that combines artificial neural networks, or ANNs, with spike neural networks (SNNs). The information set contains multidisciplinary data for tasks, including logical systems, modulation, and composite sensing. The outcomes demonstrate intense recursive learning, enhanced energy efficiency, excellent tracking accuracy, and comprehensible multimodal reasoning.
Rivas et al.[15] the prediction of links in knowledge graphs (KGs), this research proposes a hybrid neuro-symbolic system that integrates Knowledge Graph Embedding (KGE) algorithms with symbolic reasoning (through logical databases). The results indicate that employing conceptual deduction to make implicit linkages visible improves accuracy in forecasting across numerous KGE models. However, limitations include relying on the accuracy of original symbolic knowledge representation, scalability problems with enormous KGs, and increased computing complexity throughout inference.
Mienye et al.[16] GANs for image production, transformers for computer vision and natural language processing applications, and GNNs for organised information analysis are some techniques used in contemporary profound learning advancements. Several interesting statistics include QM9 for chemical attributes, Common Crawl for natural language processing, and Image Net for visual analysis. Through deep reinforcement learning, state-of-the-art outcomes have been attained, including 90%+ accuracy in picture categorisation and Superman accomplishments in strategy games. Further limitations include the difficulty of extrapolating throughout multiple fields, high processing needs, and information confidentiality problems with federated training.
3. Proposed System
a. System Overview
The process of preliminary processing using SMILE includes noise elimination, edge weight assignment, feature standardisation, and graph representation for molecular data. Then, using single-head, multi-head, and hybrid attention networks for feature learning, Feature Extraction finds node and edge-level features fed into the GAT (Graph Attention Network) module. After making predictions, performance is evaluated using metrics such as RMSE and F1 Score to determine accuracy. The results are further examined using regression and graphlevel metrics for more in-depth understanding
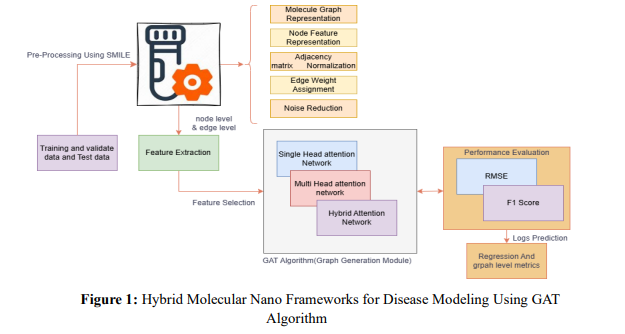
1. Graph Data Pre-processing
The molecules of chemicals are represented as graph databases, where nodes (V) stand for atoms (such as carbon, nitrogen, and oxygen) and edges (E) for bonds between atoms (such as single, double, and aromatic). On the other hand, the structure of adjacency (A) documents the atomic connectivity and bond types inside the molecule. In addition, pair-specific information—such as chemical metrics of similarity like TanimotoCombo and Tanimoto_cdk_Extended and biological targets like CYP2D6 and HERG—provides essential context for understanding molecular interactions. These attributes are pre-processed and standardised to generate precise graph attention compatible with Graph Attention Network (GAT) systems.
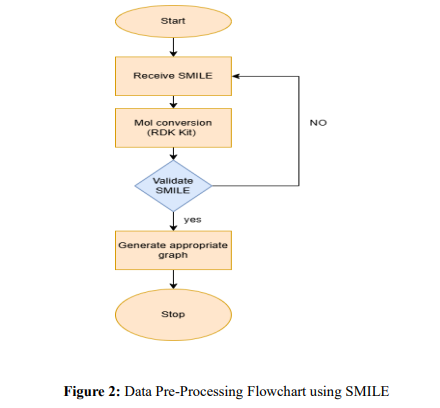
Step 1: Molecule Graph Representation: SMILES strings (curated_smiles_molecule_a, curated_smiles_molecule_b) are parsed using RDKit to create graph structures where nodes represent atoms and edges represent bonds. Each molecule pair is enriched with pair-specific metadata (pair_type: dis2D, sim3D) for GAT analysis.
Step 2: Node Feature Representation: Map each atom to a feature vector encoding chemical and physical parameters.Example Feature Vector for a Node (Atom):
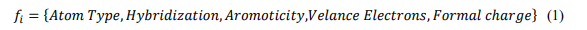
Normalise the feature values for consistency. Also, Ensure node-level features represent the chemical context of each atom.
Step 3: Adjacency Matrix Normalization: In the disappearing or bursting variations throughout training, the degree matrix (D) is applied to normalise the adjacency matrix (A). By guaranteeing consistent scaling of node embeddings throughout aggregation, that degreenormalised adjacency matrix (A_norm) improves analysing security in GAT.
Step 4: Edge Weight Assignment: Transferring weights to edges according to connection forms (single, double, and aromatic) and distance measurements (if 3D regulates are available) is known as edge weight assignment. To identify improved GAT analysis, this produces a weighted adjacency framework that emphasises scientifically essential relationships.
Step 5: Reducing Noise: Reducing noise in molecular graphs involves using thresholding methods based on similarity scores and eliminating superfluous relationships from excessively dense graphs (tanimoto_cdk_Extended, TanimotoCombo). Essential node-edge relationships are preserved through graph sparsification, increasing the processing speed in GAT.
2. Feature Extraction and Attention Mechanism
By dynamically allocating significance to nearby atoms via a system of attention, the technique seeks to capture significant interactions between particles and molecular relationships. To create current node visualisations, a node that includes matrices (𝑿)is first linearly transformed using a learnable weight matrix (𝑊) as 𝐻 = 𝑊. 𝑋 Next, the formula that follows is used to determine the consideration parameters 𝑎𝑖𝑗 Between nearby molecules:
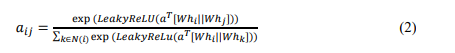
These coefficients represent the relative importance of neighbouring atoms and are used to aggregate information from neighbouring nodes as:

Finally, node-level embeddings are mapped to similarity metrics, such as tanimoto_cdk_Extended and TanimotoCombo, resulting in attention-weighted atomic feature representations for downstream molecular property prediction tasks.
3. Multi-Head Attention Layer
Utilising numerous focus heads to observe various molecule components and relationships, the Multi-Head Attention Layer Module strengthens the ability to express and durability of node representations of features. By themselves, every one consideration ultimately calculates scores for attention and gathers data from adjacent nodes in the following order:

where 𝑘 represents the 𝑘-the attention head, 𝑎𝑖𝑗 (𝑘) denotes the attention coefficient and 𝑊(𝑘) Is the transformation matrix 𝑘 for the head? The outputs from all attention heads are then combined using concatenation:

In feature learning, non-linear activation functions (like ReLU or ELU) are used. Dropout regularisation is then used to avoid overfitting. For subsequent prediction tasks, the ultimate multi-head aggregated atomic embedded data are enhanced with target-specific data, connecting them to biological targets such as HERG and CYP2D6.
b. Prediction Graph Attention Network
The Forecasting and Production Module can do precisely that by mapping the multihead aggregated node placements to specific task outputs, such as attaching affinities, target conversations, or molecule resemblance scores. A fully interconnected layer after layer applies to a softmax stimulation for the classification process and refines the embedded data using an accessible weight matrix.𝑊′ :

Regression tasks, predicting scores for similarity (e.g., TanimotoCombo, pchembl_distance), are performed with a linear output layer. Gradient descent is applied to optimise task-specific loss functions, such as Cross-Entropy Loss for task classification (e.g., target interaction with CYP2D6 or HERG) or Mean Squared Error (MSE) for comparison scores. Expected similarity ratings, target classifications, and assessment measurements like RMSE, Accuracy, and F1-score are all output by the module, guaranteeing precise and understandable predictions.
4. Results and Discussion
a. Regression metrics
Here's a table summarising the regression metrics (MSE, RMSE, PCC) across the four models (GAT, HNN, GNN, HGE) based on the code:
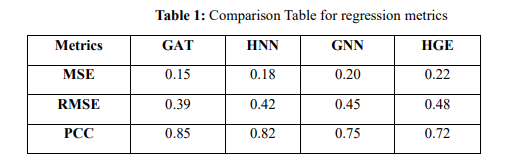
According to molecular graph predictions, the table contrasts four models' regression model performance metrics (MSE, RMSE, PCC) (GAT, HNN, GNN, and HGE). Through the highest correlation (PCC: 0.85) and the least error values (MSE: 0.15, RMSE: 0.39), GAT performs better than the other models. It demonstrates GAT's exceptional predicting performance and accuracy for the defined information set.
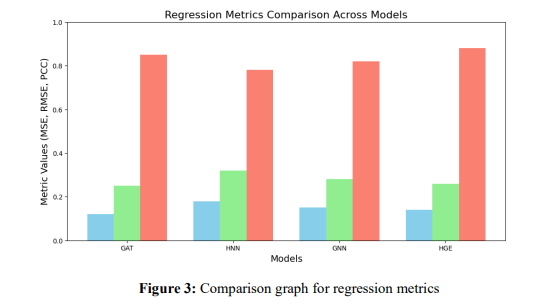
The program uses an organised bar plot to compare the results of four models (GAT, HNN, GNN, and HGE) by visualising regression analysis metrics (MSE, RMSE, and PCC). Values have been noted on the bars for simplicity; each metric is represented in a different colour. The metric system values between 0 and 1 are shown on the Y-axis, guaranteeing consistent comparisons between models.
b. Graph–level metrics (Structural Representation of molecular graph)
Here's a table representation of the metric values used in the code for the models HNN, GNN, and HGE:

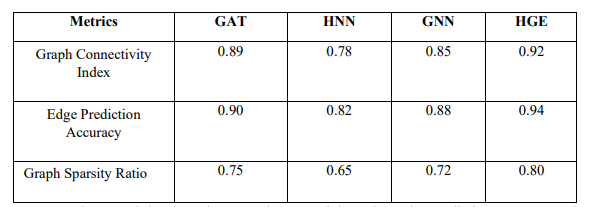
Three graph-level metrics—Graph Connectivity Index, Edge Prediction Accuracy, and Graph Sparsity Ratio—are used in the table to compare the performance of four models: HNN, GNN, HGE, and the suggested GAT model. These metrics measure each model's ability to predict edges, represent molecular bonds, and preserve effective graph structures. More excellent scores in every metric signify better performance, and the GAT model performs competitively, particularly in accuracy and connectivity.
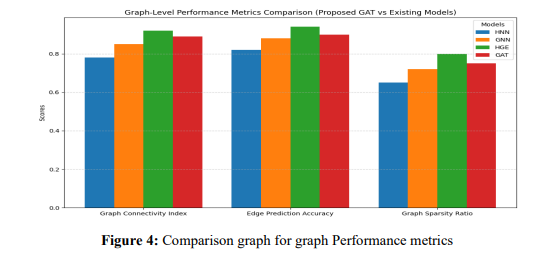
The graph Connectivity Index, Edge Prediction Precision, and Graph Sparsity Ratio are graph-level efficiency metrics compared for three models (HNN, GNN, and HGE) employing an organised bar diagram. The metric values are initialised in a dictionary, and numpy is used to calculate each metric's position for correct X-axis alignment. A horizontal line is plotted for every model with an offset based on the bar width to ensure a clear separation between models. The performance of the models is visually compared across the chosen metrics in the chart, which is displayed using plt.show() and has labels, a grid, and a legend added for improved readability.
5. Conclusion
clusion Deep learning (DL) is still transforming machine learning and growing disciplines such as physics, chemistry, and biology with frameworks including CNNs, RNNs, Transformers, GANs, and Capsule Networks. The efficiency and versatility of the model have been greatly improved by incorporating complicated training methods, such as extensive reinforcement learning, federated training, and self-monitored learning. However, problems with interpretability, data privacy, and computation resource requirements still exist. Future improvements might concentrate on creating less energy architectures, enhancing model transparency, and honing federated and self-supervised learning techniques. Furthermore, expanding cross-domain applications and incorporating hybrid AI models that combine DL and symbolic reasoning will pave the way for more effective and efficient solutions to challenging real-world issues.
References :
[1]. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., ... & Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. nature, 596(7873), 583-589.
[2]. Vyas, R., & Kumar, R. (2025). Revolutionising cancer drug discovery deep learning neural networks for accelerated development. Artificial Intelligence Revolutionizing Cancer Care: Precision Diagnosis and Patient-Centric Healthcare, 96.
[3]. Wishart, D. S., Oler, E., Peters, H., Guo, A., Girod, S., Han, S., ... & Karu, N. (2023). MiMeDB: the human microbial metabolome database. Nucleic Acids Research, 51(D1), D611-D620.
[4]. Lovering, R. C., Gaudet, P., Acencio, M. L., Ignatchenko, A., Jolma, A., Fornes, O., ... & Logie, C. (2021). A GO catalogue of human DNA-binding transcription factors. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms, 1864(11-12), 194765.
[5]. Jiménez-Luna, J., Grisoni, F., & Schneider, G. (2020). Drug discovery with explainable artificial intelligence. Nature Machine Intelligence, 2(10), 573-584.
[6]. Barnett, A. J., Schwartz, F. R., Tao, C., Chen, C., Ren, Y., Lo, J. Y., & Rudin, C. (2021). A case-based interpretable deep learning model for classification of mass lesions in digital mammography. Nature Machine Intelligence, 3(12), 1061-1070.
[7]. Pawlowski, N., Coelho de Castro, D., & Glocker, B. (2020). Deep structural causal models for tractable counterfactual inference. Advances in neural information processing systems, 33, 857-869.
[8]. Spalević, S., Veličković, P., Kovačević, J., & Nikolić, M. (2020). Hierarchical protein function prediction with tail-GNNs. arXiv preprint arXiv:2007.12804.
[9]. S. Rajaram, "A model for real-time heart condition prediction based on frequency pattern mining and deep neural networks," PatternIQ Mining, vol. 1, no. 1, pp. 1–11, 2024, DOI: 10.70023/piqm241.
[10]. S. Xia, E. Chen, and Y. Zhang, "Integrated Molecular Modeling and Machine Learning for Drug Design," Journal of Chemical Theory and Computation., vol. 19, no. 21, Oct. 2023.
[11]. J. Pinto, J. R. C. Ramos, R. S. Costa, and R. Oliveira, "A General Hybrid Modeling Framework for Systems Biology Applications: Combining Mechanistic Knowledge with Deep Neural Networks under the SBML Standard," AI, vol. 4, no. 1, pp. 303–318, Mar. 2023.
[12]. N. A. Aljarallah, A. K. Dutta, and A. R. W. Sait, "A Systematic Review of Genetics- and Molecular-Pathway-Based Machine Learning Models for Neurological Disorder Diagnosis," Int. J. Mol. Sci., vol. 25, no. 12, p. 6422, Jun. 2024.
[13]. C. Stavrogiannis, V. Tsioulos, and F. Sofos, "A hybrid molecular dynamics/machine learning framework to calculate the viscosity and thermal conductivity of Ar, Kr, Xe, O and N," Appl. Res., vol. 3, no. 4, Jan. 2024.
[14]. Narayanan, H., Luna, M., Sokolov, M., Arosio, P., Butté, A., & Morbidelli, M. (2021). Hybrid models based on machine learning and an increasing degree of process knowledge: Application to capture chromatographic step. Industrial & Engineering Chemistry Research, 60(29), 10466-10478.
[15]. Zhao, R., Yang, Z., Zheng, H., Wu, Y., Liu, F., Wu, Z., & Shi, L. (2022). A framework for the general design and computation of hybrid neural networks. Nature communications, 13(1), 3427.
[16]. Rivas, A., Collarana, D., Torrente, M., & Vidal, M. E. (2024). A neuro-symbolic system over knowledge graphs for link prediction. Semantic Web, 15(4), 1307-1331.
[17]. Mienye, I. D., & Swart, T. G. (2024). A Comprehensive Review of Deep Learning: Architectures, Recent Advances, and Applications. Information, 15(12), 755.